
A pesquisa na área de materiais tem tido grande avanço nos últimos anos, indo de um enfoque basicamente aplicado e relacionado à engenharia para uma posição onde tem grande impacto em outras áreas, incluindo a física, química e biologia. Neste sentido, pesquisas teóricas e computacionais vêm ganhando muito espaço, principalmente devido à grande capacidade preditiva destas metodologias. Entre as metodologias mais utilizadas, destacam-se os métodos baseados na mecânica quântica, principalmente aqueles baseados na Teoria do Funcional da Densidade, metodologia que deu o Prêmio Nobel de Química a Walter Kohn em 1998.[1] Hoje este tipo de metodologia chegou à sua maturidade, tendo garantido os princípios da reprodutibilidade e confiabilidade de diversos códigos computacionais, conforme publicado recentemente na revista Science.[2]
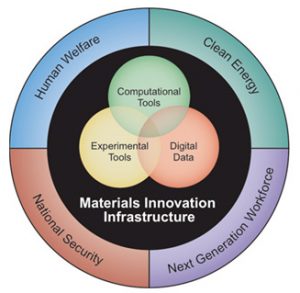
 A comunidade brasileira já possui habilidades que os coloca em destaque na comunidade mundial quando o assunto se refere à ciência computacional de materiais. Artigos de grupos paulistas já veicularam as capas de inúmeras revistas de impacto, e inúmeros trabalhos se tornaram referência mundial. Hoje, no estado de São Paulo, há cerca de 20 grupos de pesquisa trabalhando com essas metodologias. Alguns deles já figuraram nas capas das mais importantes revistas científicas internacionais, como pode ser visto na imagem ao lado. Uma vez que a área já atingiu sua maturidade, é preciso estar atento às contínuas inovações do setor, a fim de que nossa comunidade se mantenha atualizada e na vanguarda do conhecimento. Uma ação de grande peso neste sentido ocorreu quando o governo americano lançou o programa “Materials Genome”, que visava criar uma nova era na descoberta e manufatura de materiais, aumentando significativamente a velocidade de descoberta dos materiais e mantendo o seu custo a uma fração do que é hoje. O diagrama ao lado indica os principais conceitos com relação a essa iniciativa.
A comunidade brasileira já possui habilidades que os coloca em destaque na comunidade mundial quando o assunto se refere à ciência computacional de materiais. Artigos de grupos paulistas já veicularam as capas de inúmeras revistas de impacto, e inúmeros trabalhos se tornaram referência mundial. Hoje, no estado de São Paulo, há cerca de 20 grupos de pesquisa trabalhando com essas metodologias. Alguns deles já figuraram nas capas das mais importantes revistas científicas internacionais, como pode ser visto na imagem ao lado. Uma vez que a área já atingiu sua maturidade, é preciso estar atento às contínuas inovações do setor, a fim de que nossa comunidade se mantenha atualizada e na vanguarda do conhecimento. Uma ação de grande peso neste sentido ocorreu quando o governo americano lançou o programa “Materials Genome”, que visava criar uma nova era na descoberta e manufatura de materiais, aumentando significativamente a velocidade de descoberta dos materiais e mantendo o seu custo a uma fração do que é hoje. O diagrama ao lado indica os principais conceitos com relação a essa iniciativa.
 Uma das principais ações neste sentido tem sido a criação de grandes repositórios de dados sobre inúmeros materiais, obtidos através de simulações de primeiros princípios. Para isso, faz-se uso de robôs que varrem a tabela periódica juntando elementos em diferentes estruturas cristalinas, e calculando propriedades básicas destes materiais. A maioria dos materiais simulados ainda não foram estudados e, algumas vezes, nem são estáveis. A quantidade de dados disponíveis é gigantesca. Algumas das iniciativas possuem mais de 1 milhão de materiais simulados. Esse tipo de iniciativa foi, inclusive, capa de uma das edições da Revista Nature no mês de Maio de 2016.[3] Com o advento destes repositórios, a área de simulação computacional de materiais deve tomar um novo rumo, onde o foco do estudo não será mais descobrir as propriedades de um material em particular, mas passa a ser o de buscar quais os materiais que possuem uma a propriedade desejada para uma aplicação específica.
Uma das principais ações neste sentido tem sido a criação de grandes repositórios de dados sobre inúmeros materiais, obtidos através de simulações de primeiros princípios. Para isso, faz-se uso de robôs que varrem a tabela periódica juntando elementos em diferentes estruturas cristalinas, e calculando propriedades básicas destes materiais. A maioria dos materiais simulados ainda não foram estudados e, algumas vezes, nem são estáveis. A quantidade de dados disponíveis é gigantesca. Algumas das iniciativas possuem mais de 1 milhão de materiais simulados. Esse tipo de iniciativa foi, inclusive, capa de uma das edições da Revista Nature no mês de Maio de 2016.[3] Com o advento destes repositórios, a área de simulação computacional de materiais deve tomar um novo rumo, onde o foco do estudo não será mais descobrir as propriedades de um material em particular, mas passa a ser o de buscar quais os materiais que possuem uma a propriedade desejada para uma aplicação específica.
Há cerca de uma dezena de iniciativas como essa no mundo, e todas ainda estão em um estágio bastante inicial. A área ainda busca sua estruturação e disseminação, e isso significa uma grande oportunidade para tornar São Paulo em um importante parceiro dessas iniciativas. Entre as principais iniciativas estão o NoMad,[4] liderado pelo Físico alemão Mathias Scheffler, o Aflow,[5] da Universidade de Duke, nos Estados Unidos, e o Materials Project,[6] iniciativa conjunta entre Berkeley e o MIT. As possibilidades de aplicações são muito amplas, passando pela busca de materiais para células solares, materiais termoelétricos, catálise, baterias, materiais magnéticos, nanoeletrônica entre outros.
Uma das aplicações recentes remete aos materiais conhecidos como TCOs (óxidos condutores transparentes), que são usados, por exemplo, em janelas inteligentes. Hoje o material mais utilizado e mais estudado para este fim é o ITO (Indium Tin Oxide). A pergunta a ser feita é: “Quais outros materiais podem ser utilizados para esse tipo de aplicação?”. Ao invés de ir ao laboratório e crescer todos materiais via ‘tentativa e erro’, podemos varrer os bancos de dados procurando materiais com um gap de energia específico e com massas efetivas específicas, que estejam dentro dos valores desejados para a aplicação. Uma vez encontrados esses materiais, que podem estar fora da família do ITO, leva-se essa informação ao laboratório para que os materiais sejam produzidos e testados.[7] Outra aplicação remete a materiais termoelétricos. A busca por melhores termoelétricos tem sido um dos principais focos desta comunidade. A estratégia tem sido sempre a mesma: crescer um material, medir suas propriedades e verificar se são adequadas. Isso gera um grande custo com baixa eficiência. Com o Big Data, pode-se varrer os bancos de dados procurando materiais com Figura de Mérito específica, e sugerir que esses materiais sejam investigados e crescidos. Este approach é muito mais rápido e barato do que o outro.[8]
Uma vez estabelecida esta nova rota de descoberta e design de materiais no cenário internacional, é imprescindível que a comunidade brasileira também passe a atuar nesta frente. Estrategicamente é interessante que pesquisadores do estado de SP façam parte das iniciativas mundiais já começadas, atuando como parceiros na construção dos bancos de dados e se especializando na busca de materiais, além, certamente, de dominarem as metodologias inerentes a esta área. Além do conhecimento relacionado à área de Física e Ciência dos Materiais, há muito a ser desenvolvido com respeito a Ciência da Computação, incluindo algoritmos otimizados de busca, machine learning e data mining.
Uma iniciativa mais ampla nesta área trará benefícios diretos para os pesquisadores brasileiros, mas também tem potencial de alavancar a competitividade de empresas inovadoras, já que oferece rotas fora do padrão para a busca de novos materiais. O Programa de Pós-Graduação em Nanociências e Materiais Avançados da UFABC avança nessa direção, e seus pesquisadores já desenvolvem projetos na área.
[1] W. Kohn, Nobel Lecture: Electronic structure of matter—wave functions and density functionals, Reviews of Modern Physics 71, 1253 (1999).
[2] K. Lejaeguere, et al., Reproducibility in density functional theory calculations of solids, Science 351, 1394 (2016).
[3] N. Nosengo, The material code, Nature, 533, 23 (2016).
[4] http://nomad-repository.eu/cms/
[5] http://www.aflowlib.org/
[6] https://materialsproject.org/
[7] G. Hautier, et al., Identification and design principles of low hole effective mass p-type transparent conducting oxides, Nature Comm. 4, 2291 (2013).
[8] S .Curtarolo, et al., The high-throughput highway to computational materials design, Nature Mat. 12, 191 (2013).