Simulações computacionais são importantes ferramentas para a área, visto que podem explicar inúmeros fenômenos observados experimentalmente, bem como propor novos fenômenos e novos experimentos. Diversos projetos de pesquisa vinculados ao Programa de Pós-Graduação em Nanociências e Materiais Avançados focam no estudo de materiais de um ponto de vista computacional. A UFABC possui uma grande infraestrutura computacional para dar suporte a estes pesquisadores e seus alunos, incluindo um data center dedicado com mais de 3000 processadores disponíveis para utilização. Os trabalhos desenvolvidos cobrem diversos temas distintos, indo desde o estudo de materiais cristalinos, passando por materiais bidimensionais (2D), e chegando a modelagem de processos bioquímicos complexos importantes que ocorrem dentro das células humanas. Realmente, a nanotecnologia tem aplicações que ultrapassam as fronteiras dos materiais convencionais.

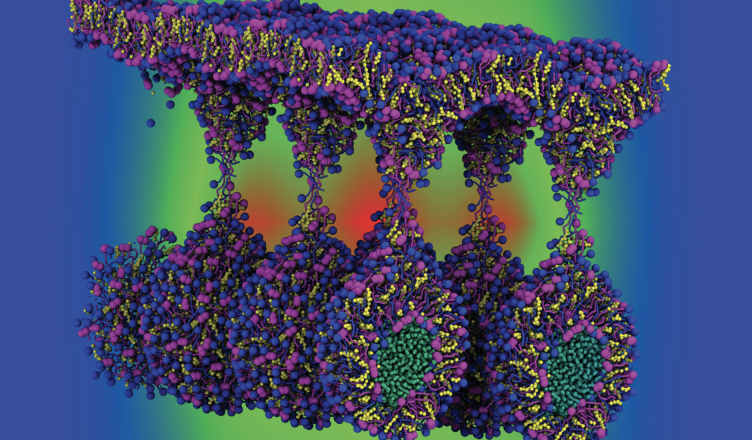
As nanoemulsões lipídicas são nanomateriais promissores para aplicações de entrega de medicamentos nas indústrias alimentícia, farmacêutica e cosmética. Apesar do notável interesse comercial, pouco se sabe sobre sua organização supramolecular, especialmente sobre como tais formulações multicomponentes interagem com as membranas celulares. No presente trabalho, simulações de dinâmica molecular de granulação grossa foram empregadas para estudar a automontagem de uma gotícula de nanoemulsão lipídica de 15 componentes contendo vitaminas A e E para liberação na pele.
Nossos resultados exibem aspectos de aglomeração “semelhante a cebola” entre os constituintes químicos nas diferentes camadas da nanogotícula lipídica.
As moléculas de vitamina E estão mais concentradas no centro da gota junto com outros constituintes hidrofóbicos, como os triglicerídeos de cauda longa. Por outro lado, a vitamina A ocupa uma camada intermediária entre o núcleo e a superfície coemulsificante da nanogotícula, juntamente com os fosfolipídios de lecitina.
Simulações de dinâmica molecular de granulação grossa também foram realizadas para fornecer informações sobre os primeiros passos envolvidos na absorção e penetração da nanogota através de modelos de membrana da pele, representando uma via intracelular (infundíbulo do folículo piloso) e intercelular (estrato córneo) através da pele.
Nossos dados fornecem uma primeira visão sobre a complexa organização da nanoemulsão comercial e sua interação com as membranas da pele. Esperamos que nossos resultados abram o caminho para o design racional de tais nanomateriais.
https://pubs.rsc.org/en/content/articlelanding/2022/nr/d1nr04610a
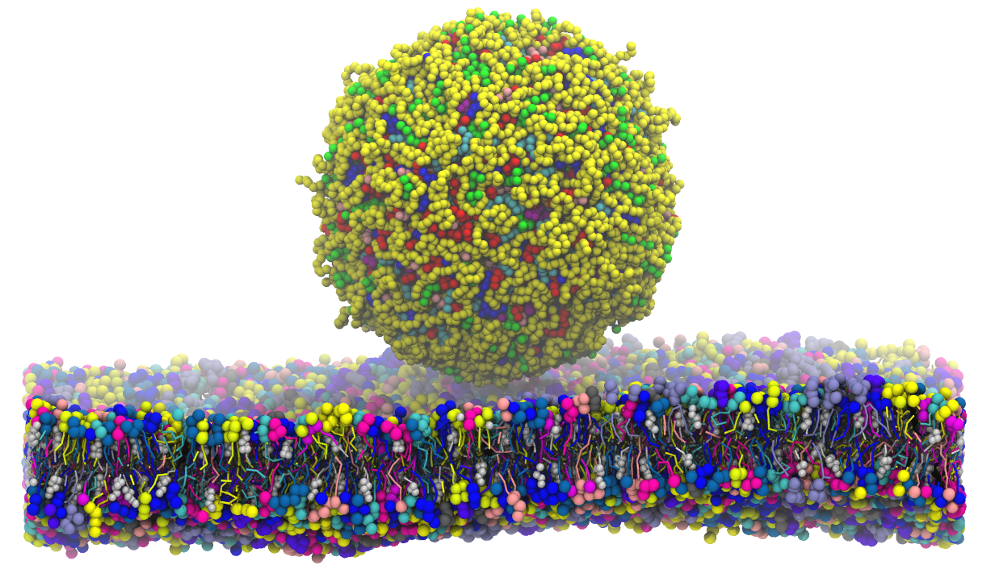
A busca por medicamentos e terapias mais personalizadas, que minimizem efeitos colaterais é uma área muito ativa de pesquisa atualmente. Uma abordagem muito investigada envolve desenvolver medicamentos que possam ser absorvidos pela pele, acessando rapidamente a corrente sanguínea. Há um grande desafio neste caso: vencer a pele! É uma barreira natural que, dentre suas várias funções, trabalha para impedir a entrada de compostos estranhos.
Neste trabalho utilizando simulações computacionais de dinâmica molecular foi construído um modelo de pele (> 60 mil átomos) para se estudar condições onde seria possível facilitar a hidratação da pele. Observamos que ao aplicar um campo elétrico externo constante, vesículas contendo água se formavam, atravessando a barreira. Olha que interessante … medicamentos solúveis em água já poderiam pegar uma carona aí.
Trabalho destaque escolhido como uma das capas na edição da revista.

 O controle da permeabilidade da pele a substâncias específicas (por exemplo, medicamentos, vitaminas e nutrientes) através do estrato córneo é um desafio. A iontoforese é uma opção, apesar da falta de uma compreensão detalhada do mecanismo molecular subjacente.
O controle da permeabilidade da pele a substâncias específicas (por exemplo, medicamentos, vitaminas e nutrientes) através do estrato córneo é um desafio. A iontoforese é uma opção, apesar da falta de uma compreensão detalhada do mecanismo molecular subjacente.
No presente trabalho, as simulações referentes à aplicação de um campo elétrico externo contínuo ao estrato córneo, em uma faixa de baixa intensidade (0–24 mV/nm-1), foram realizadas utilizando a abordagem de dinâmica molecular de granulação grossa. Usando um conjunto de réplicas de sementes aleatórias da configuração inicial, observamos que na faixa de intensidade do campo elétrico de 22-23 mV/nm-1, vesículas lipídicas ricas em água foram formadas em 20% dos casos. Poros apareceram nos 80% restantes. Argumentamos que os lipídios sofrem rápidas reorientações sob campo elétrico, induzindo instabilidade mecânica, que dá origem aos poros.
Apresentamos um modelo eletrostático simples para interpretar os resultados onde a incompatibilidade entre as permissividades elétricas da membrana e meios externos e o gradiente do campo elétrico local na superfície da membrana governam as escalas de tempo e os campos elétricos para a formação de vesículas.
Nossos resultados indicam que apenas 10% de diferença entre as permissividades elétricas da membrana e da mídia externa diminui 1/6 do tempo mínimo necessário para a formação da vesícula. O campo elétrico mínimo necessário diminui 10 vezes.
O controle e sintonia da formação de vesículas biologicamente compatíveis, capazes de transportar substâncias sob campos elétricos de baixa intensidade, tem uma aplicação promissora em áreas como a farmacoterapia e dermocosméticos permitindo o uso de substâncias hidrofílicas em aplicações dérmicas.
Acesse todos os detalhes em: https://pubs.acs.org/doi/full/10.1021/acs.nanolett.9b03881
Pela primeira vez, nanocompósitos de poliuretano termoplástico (TPU) + MXene são investigados pela DFT. O efeito de diferentes grupos de terminação de superfície MXene na adsorção de TPU é investigado e os mecanismos de interações TPU-MXenes são descritos.

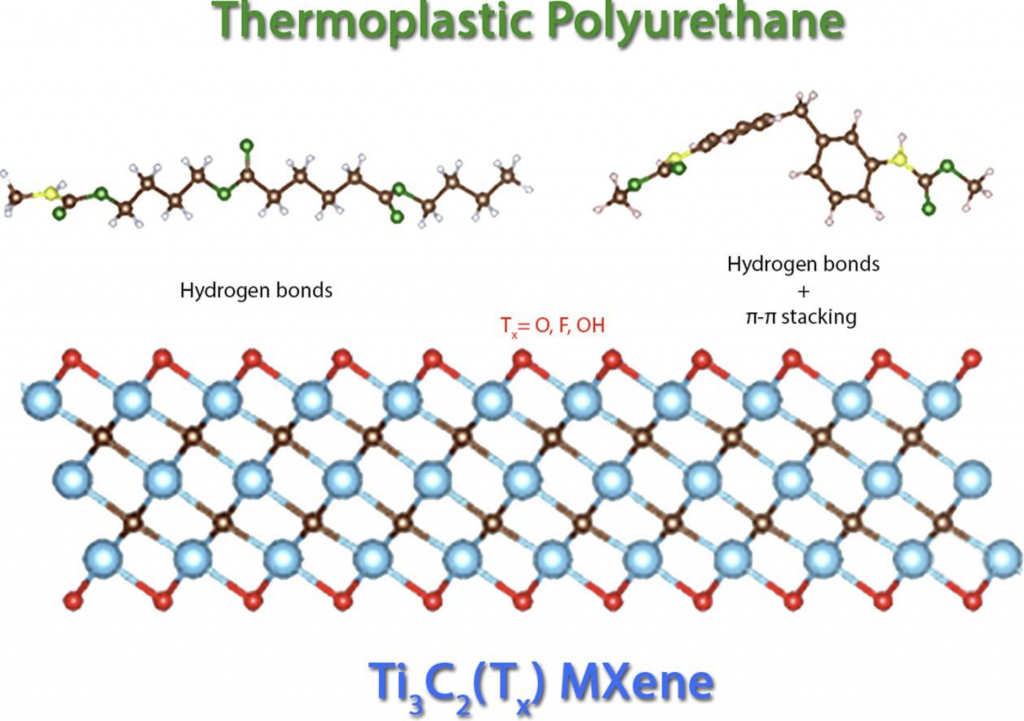 Os MXenes recentemente descobertos são candidatos promissores como reforços para nanocompósitos devido às suas altas propriedades mecânicas, condutividades térmicas e elétricas. Essas propriedades são fortemente afetadas pela presença de grupos funcionais de superfície (
Os MXenes recentemente descobertos são candidatos promissores como reforços para nanocompósitos devido às suas altas propriedades mecânicas, condutividades térmicas e elétricas. Essas propriedades são fortemente afetadas pela presença de grupos funcionais de superfície (  O, F ou OH), que estão relacionados à rota de síntese empregada. No entanto, faltam investigações científicas sobre a influência de tais grupos funcionais na interação MXene / matriz polimérica. Portanto, realizamos cálculos de teoria funcional de densidade para simular a interação entre Ti 3 C 2MXenes com diferentes grupos funcionais e moléculas à base de poliuretano termoplástico (TPU). Verificou-se que os principais mecanismos de interação envolvidos foram a formação de ligações de hidrogênio e o empilhamento π-π (no anel aromático). Além disso, enquanto as terminações de flúor e hidroxila favoreciam a interação com TPU, o MXene terminado em oxigênio a impediu em três das quatro configurações testadas. Essas descobertas indicam a relevância do controle da química de superfície MXenes para melhorar as interações MXenes / matrizes poliméricas em nanocompósitos.
O, F ou OH), que estão relacionados à rota de síntese empregada. No entanto, faltam investigações científicas sobre a influência de tais grupos funcionais na interação MXene / matriz polimérica. Portanto, realizamos cálculos de teoria funcional de densidade para simular a interação entre Ti 3 C 2MXenes com diferentes grupos funcionais e moléculas à base de poliuretano termoplástico (TPU). Verificou-se que os principais mecanismos de interação envolvidos foram a formação de ligações de hidrogênio e o empilhamento π-π (no anel aromático). Além disso, enquanto as terminações de flúor e hidroxila favoreciam a interação com TPU, o MXene terminado em oxigênio a impediu em três das quatro configurações testadas. Essas descobertas indicam a relevância do controle da química de superfície MXenes para melhorar as interações MXenes / matrizes poliméricas em nanocompósitos.
Para mais detalhes acesse https://www.sciencedirect.com/science/article/pii/S0169433220312836
Os avanços na síntese e controle de crescimento em nanoescala permitiram explorar uma variedade de materiais bidimensionais (2D) [1]. Esses sistemas mostram propriedades eletrônicas, de transporte e mecânicas muito exclusivas [2, 3]. De particular interesse são os isoladores topológicos 2D [4], apesar da realização experimental em poços quânticos de HgTe / CdTe [5] mais de uma década atrás, foram observados em muito poucos sistemas até agora [6]. Neste trabalho, com base em uma modelagem realista de materiais em nanoescala, propomos um novo caminho para investigar isoladores topológicos 2D usando materiais amorfos.


As propriedades topológicas dos materiais estão, até agora, associadas às características de sua estrutura cristalina, embora a simetria translacional não seja um requisito explícito das fases topológicas. Estudos recentes de modelos de salto em treliças aleatórias demonstraram que os sistemas de modelos amorfos mostram uma topologia não trivial. Usando cálculos ab initio , mostramos que os materiais amorfos bidimensionais também podem exibir propriedades topológicas do isolador. Mais especificamente, apresentamos um estudo realista e avançado das propriedades eletrônicas e de transporte dos sistemas de bismuteno amorfo, mostrando que esses materiais são isoladores topológicos. Esses sistemas são caracterizados pelo índice topológico 2 = 1 e pela dualidade de borda grossa, e sua condutância linear é quantizada,
 , para energias Fermi dentro da lacuna topológica. Nosso estudo abre o caminho para a investigação experimental e teórica de materiais isolantes topológicos amorfos.
, para energias Fermi dentro da lacuna topológica. Nosso estudo abre o caminho para a investigação experimental e teórica de materiais isolantes topológicos amorfos.
Leia o artigo na integra em https://pubs.acs.org/doi/abs/10.1021/acs.nanolett.9b03881 e acesse as referências numéricas.
 Os nanocristais (CNs) apresentam propriedades físico-químicas únicas decorrentes do tamanho e da presença de ligantes. Compreender e controlar as interações ligante-cristal, bem como o processo de troca de ligantes, é um dos temas centrais na ciência da NC atualmente. No entanto, a relação entre desordem estrutural da NC e o efeito da troca de ligantes na estrutura atômica da NC ainda não é suficientemente compreendida.
Os nanocristais (CNs) apresentam propriedades físico-químicas únicas decorrentes do tamanho e da presença de ligantes. Compreender e controlar as interações ligante-cristal, bem como o processo de troca de ligantes, é um dos temas centrais na ciência da NC atualmente. No entanto, a relação entre desordem estrutural da NC e o efeito da troca de ligantes na estrutura atômica da NC ainda não é suficientemente compreendida.
Neste trabalho foi combinado a análise da função de distribuição de pares a partir de dados de difração de elétrons, estrutura fina de absorção de raios X estendida e microscopia eletrônica de transmissão de alta resolução como técnicas experimentais e cálculos da teoria funcional da densidade dos primeiros princípios para elucidar os efeitos da troca de ligantes na estrutura ZrO 2 NC .
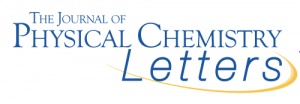

Acesse aqui https://pubs.acs.org/doi/10.1021/acs.jpclett.9b00439 e leia o estudo completo. (DOI: https://doi-org.ez42.periodicos.capes.gov.br/10.1021/acs.jpclett.9b00439)
Um grande número de estruturas topolóides exóticas foi recentemente previsto pela pesquisa de compostos cujos pontos de passagem calculados da estrutura da banda atendem a requisitos específicos de simetria. A descoberta de fenômenos físicos emocionantes por estudos experimentais desses compostos previstos está chegando.

No entanto, o exame de algumas dessas estruturas assumidas de alta simetria sugere que nem sempre a montagem de átomos em uma configuração que produz propriedades topológicas exóticas seja protegida contra a quebra de simetria de redução de energiamodos. De fato, embora as características topológicas em massa levem a estados de superfície / borda protegidos, nada protege os estados em massa da instabilidade estrutural. O ônus da prova para previsões teóricas de fenômenos físicos empolgantes deve incluir algumas dicas convincentes de que tais fenômenos podem viver em compostos termodinamicamente estáveis (ou quase estáveis). Aqui, ilustramos como o uso das energias totais calculadas (elétron + íon) das estruturas candidatas pode remover topoloides previstos falso-positivos da lista de compostos possíveis de realizar, em benefício do muito apreciado processo iterativo de descoberta de materiais teórico-experimentais.
Leia o artigo na integra em https://www.sciencedirect.com/science/article/pii/S1369702119307412
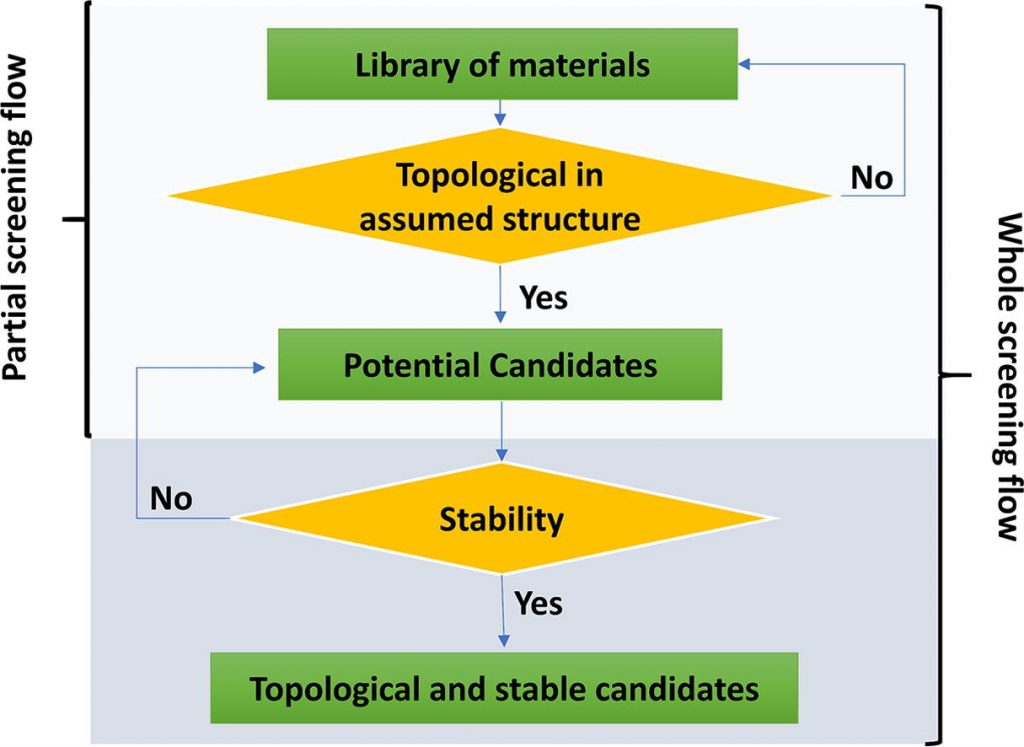
As interfaces água-mineral são importantes para vários processos ambientais, industriais, biológicos e geológicos. Gesso, CaSO 4 · 2H 2O, é um mineral generalizado de alta relevância tecnológica, médica e ambiental, mas pouco se sabe sobre sua estrutura superficial e sua interação com a água. Um entendimento em nível molecular da interface gesso / água é apresentado aqui por um estudo experimental / teórico combinado.

Investigamos a estrutura e dinâmica da água adsorvida pelo vapor na superfície de cristal único de gesso (010) à temperatura ambiente, combinando experimentos de espectroscopia vibracional de geração de soma e frequência (SFG) e simulações de dinâmica molecular inicial (AIMD). Os espectros SFG de gesso a baixa umidade relativa (UR) mostram um arranjo anisotrópico de moléculas estruturais de água e a presença de grupos OH oscilantes. As simulações AIMD permitem uma atribuição detalhada dos espectros SFG e mostram que a superfície clivada (010) se rearranja para ter apenas 25% dos grupos OH apontando para fora da superfície. Em URs mais altas, a primeira camada de água adsorvida se liga a esses grupos OH e forma um arranjo anisotrópico, mas com a quantidade de grupos OH livres suprimida significativamente e sem difusão significativa. Após a adsorção de uma segunda camada de água, embora a camada superior de moléculas seja mais desordenada e dinâmica do que a anterior, sua estrutura ainda é influenciada pela superfície de gesso por baixo, porque possui uma quantidade muito reduzida de grupos OH livres em relação à superfície livre da água e uma difusão mais lenta da superfície em relação à água a granel. Os resultados teóricos corroboram os experimentais e fornecem uma caracterização atômica precisa da estrutura da superfície.
Clique e acesse o artigo completo https://pubs.acs.org/doi/abs/10.1021/jacs.8b09907
 As metodologias usadas para procurar e desenvolver novos materiais evoluíram nas últimas décadas a partir de uma abordagem edisoniana de tentativa e erro em direção a abordagens inteligentes para descoberta e design de materiais. Espera-se uma mudança ainda mais rápida nos próximos anos, com o crescente uso da inteligência artificial em muitas áreas da química e da ciência dos materiais. Esta edição da ACS Applied Materials & Interfaces apresenta um fórum sobre descoberta e design de materiais, destacando uma amostra de trabalhos recentes em campo.
As metodologias usadas para procurar e desenvolver novos materiais evoluíram nas últimas décadas a partir de uma abordagem edisoniana de tentativa e erro em direção a abordagens inteligentes para descoberta e design de materiais. Espera-se uma mudança ainda mais rápida nos próximos anos, com o crescente uso da inteligência artificial em muitas áreas da química e da ciência dos materiais. Esta edição da ACS Applied Materials & Interfaces apresenta um fórum sobre descoberta e design de materiais, destacando uma amostra de trabalhos recentes em campo.


Para mais informações acesse https://pubs.acs.org/doi/full/10.1021/acsami.9b10631

Simulação de novos materiais autorregenerativos é destaque da edição do Journal of Computational Chemistry
Materiais inteligentes, cujas propriedades e respostas podem ser projetadas de acordo com estímulos externos, podem ser aplicados nas mais diversas áreas, desde biomateriais até na engenharia aeroespacial. Dentre os novos materiais inteligentes, os materiais autorregenerativos têm uma posição de destaque devido à sua capacidade de regenerar como resposta às perturbações externas ou condições ambientais, como luz, mudança de pH e calor. Assim, podem levar à prevenção e até mesmo gerenciamento de danos que ocorrem em várias aplicações.
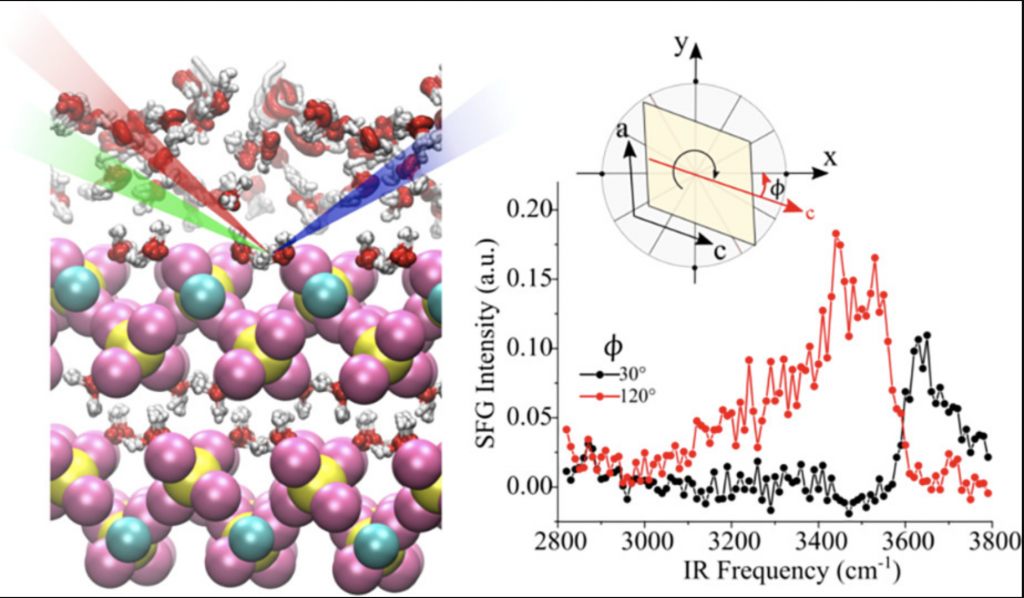
A regeneração autônoma pode ser alcançada em materiais poliméricos por diferentes mecanismos; uma possibilidade (chamada regeneração intrínseca) é o uso de ligações dinâmicas, uma classe de ligações que podem quebrar e reformar seletivamente em condições de equilíbrio. A obtenção de um material que pode autorregenerar em condições ambiente é uma grande promessa e desafio na ciência dos materiais inteligentes. A ligação covalente dinâmica da diarilbibenzofuranona (DABBF) é uma solução promissora, mas caso reaja com oxigênio não apresentará a autorregeneração.
Na página 2675 do volume 38, edição 31, G.R. Schleder, J.T. Arantes e A. Fazzio estudaram a formação da ligação dinâmica do DABBF contra a oxidação usando a teoria do funcional da densidade (DFT), mostrando que a reação de autorregeneração é favorecida.
A imagem da capa mostra o DABBF, oxigênio, arilbenzofuranona (ABF) e seus orbitais de fronteira.
O estudo completo pode ser acessado em https://doi.org/10.1002/jcc.24899. (DOI: 10.1002/jcc.24899)
Em um outro trabalho recente, o grupo do Prof. Gustavo M. Dalpian estudou as propriedades de fases deficientes em oxigênio do TiO2, observando que este material seria potencialmente interessante para a armazenagem de cargas. Esse tipo de material é encontrado usualmente nos memoristores, que são novos dispositivos eletrônicos capazes de armazenar informação em seu estado de resistência. Uma reportagem completa acerca desse interessante dispositivo fora realizadas pela Fapesp na edição 247 de Setembro 2016.
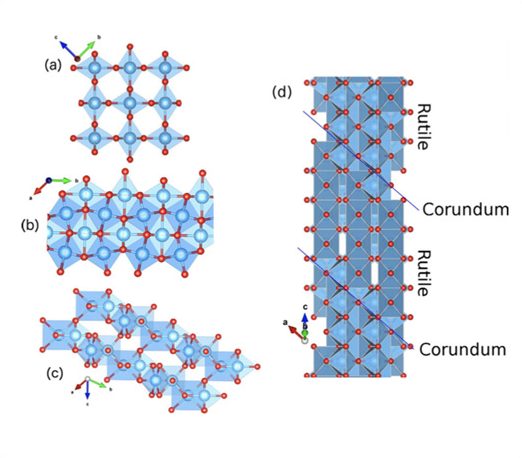
Em trabalho publicado na revista Scientific Reports do grupo Nature, o grupo demonstrou que esses materiais, conhecidos como fases Magnéli do TiO2, possuem uma banda intermediária que possui características semelhantes àquelas de níveis de defeito, podendo então ser usados para o armazenamento de carga. Na figura abaixo, mostramos a localização destas cargas quando adicionadas ao material.
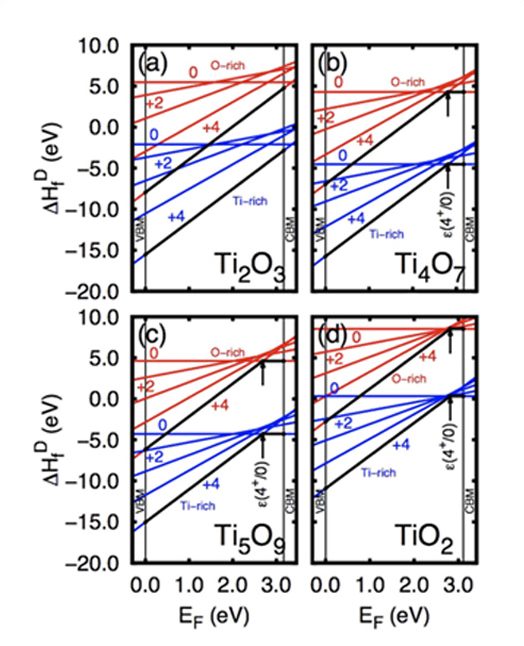
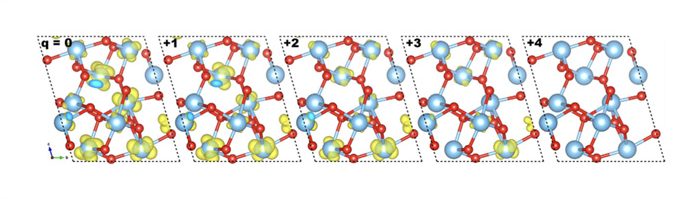
Leia o artigo na integra em Charge storage in oxygen deficient phases of TiO2: defect Physics without defect
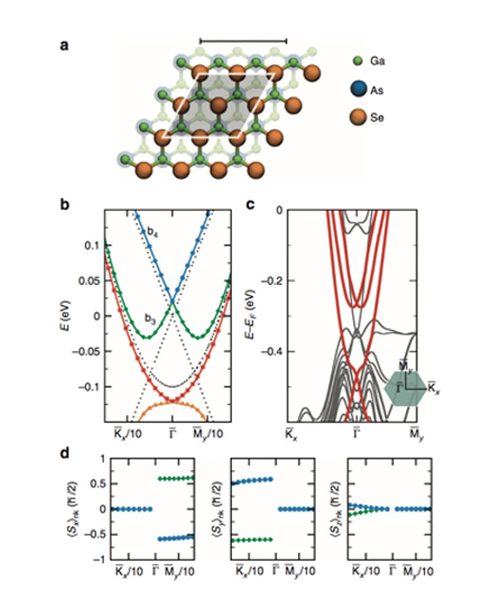
De acordo com simulações realizadas por físicos da Universidade de São Paulo (USP) e do Instituto Politécnico Rensselaer, nos Estados Unidos um novo tipo de material especial, capaz de conduzir eletricidade em sua superfície, não em seu interior, poderia ganhar versatilidade – e conduzir eletricidade em várias direções e com níveis de energia diferentes – após ser colocado em contato com um material semicondutor de eletricidade usado há décadas em computadores, foi descoberto. O trabalho apresentando esse sistema e suas propriedades interessantes não só para o entendimento do mecanismo de transporte elétrico, mas também para aplicações tecnológicas foi publicado na revista Nature Communications.

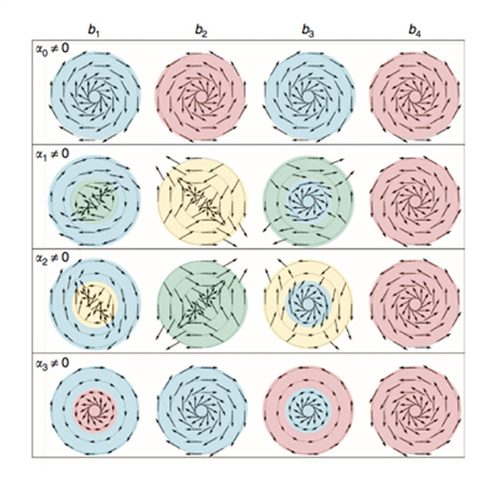
Direções possíveis dos spins da corrente elétrica: As setas indicam os vários sentidos dos spins na superfície de contato entre o arseneto de gálio e o seleneto de bismuto, como resultado da interação entre os materiais. Cada círculo representa níveis diferentes de energia dos elétrons.